
Molecular Tweezers
Novel drug candidates
For over 15 years, we have been developing Molecular Tweezers as novel drug candidates for amyloidoses and related conditions. These compounds were invented by Professors Frank-Gerrit Klärner and Thomas Schrader at the University of Duisburg-Essen, Germany, and our group discovered their ability to prevent abnormal protein aggregation. We have been collaborating with multiple groups at UCLA and around the world on these development efforts.
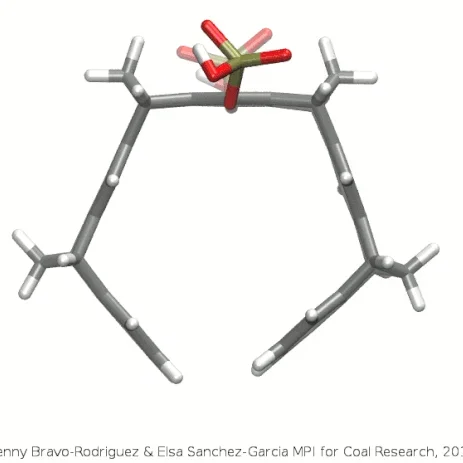
A schematic three-dimensional structure of the molecular tweezers CLR01. C atoms are represented in gray, H, in white, P in gold, and O in red.
The early days of investigation focused on characterizing the effect of the molecular tweezers on the aggregation and toxicity of Aβ. Later, this investigation was expanded to other amyloidogenic proteins leading to a 2011 paper demonstrating the inhibitory effect of a lead molecular tweezer, called CLR01, on 10 different proteins. In follow-up work, we compared CLR01 to two compounds predicted to have similar effects, which have gone on to clinical trials – the sugar derivative scyllo-inositol, and the green-tea polyphenol EGCG. CLR01 was found to have similar activity to EGCG in vitro and in cell culture and both compounds performed better than scyllo-inositol in the tests we used (Sinha et al., 2012). Mass-spectrometry experiments by Joe Loo’s group at UCLA, and NMR experiments by Chunyu Wang’s group at Rensselaer Polytechnic Institute showed that CLR01 bound to Aβ at the expected binding sites – the lysine and arginine residues, whereas no clear binding site was found for EGCG on Aβ, leaving the mechanism of action of EGCG unclear (for a detailed comparison of CLR01 and EGCG, see our 2013 review. For more general reviews about the molecular tweezers’ activity see Attar and Bitan 2014, Schrader et al., 2016, Hadrovic et al., 2019, and Shahpasand-Kroner et al., 2023)
The main binding sites of CLR01 are lysine residues, and the binding of CLR01 to lysines disrupts molecular interactions that are important for the abnormal self-assembly of different proteins, which leads to the formation of toxic oligomers and aggregates. To test the importance of lysine residues in Aβ, we studied Aβ derivatives in which each of the two lysines was substituted by an alanine (a small amino acid that cannot form the same types of molecular interactions as the larger lysine). We found not only that both the lysines were important mediators of the aggregation of Aβ, but also that one of them, in particular, was crucial for Aβ toxicity. When this particular lysine – at position 16 in the Aβ amino acid sequence – was replaced by alanine, the toxicity almost completely disappeared!
Important questions regarding the mechanism of action of the molecular tweezers are whether they enter into cells and if they do, do they localize in particular cellular compartments. This question was difficult to answer for several years because the intrinsic fluorescence of CLR01 and similar derivatives is too weak to observe on the background of a cell. Later, the generation of fluorescently labeled derivatives by the Schrader group enabled addressing these questions. The fluorescent derivatives were found to be internalized quickly by neurons, astrocytes, and oligodendrocytes (Li et al., Commun. Biol. 2021, Hererra-Vaquero et al., Biochim. Biophys. Acta Mol. Basis Dis., 2019). The main mechanism of entry was shown to be dynamin-mediated internalization. Moreover, the molecular tweezers were found to have a preference for accumulation in acid compartments, primarily lysosomes, and to a lower extent late endosomes and autophagosomes (Li et al., Commun. Biol. 2021). These findings have important implications for lysosomal storage diseases, such as mucopolysaccharidoses (see below).
Alzheimer’s-disease-related studies: Although in advanced Alzheimer’s disease the brain suffers massive atrophy, the earliest stages of the disease are characterized by a loss of synapses. At that stage, neurons are still alive, but they fail to communicate. This manifests as a loss of tiny protrusions on the nerve cells, called dendritic spines, and a decline in the electrophysiologic activity of the affected neurons. Using fluorescence microscopy, we showed that Aβ42 oligomers caused a sharp decline in the number of dendritic spines on neurons and that this toxic effect of Aβ42 could be prevented by CLR01. In addition, experiments by Claudio Grassi’s group at the Catholic University in Rome, Italy, showed that CLR01 prevented the decline in electrical activity by the neurons (Attar et al., 2012).
We then studied the effect of CLR01 in a mouse model of Alzheimer’s disease in collaboration with the Frautschy group at UCLA and showed that following treatment with just 40 micrograms per kilogram a day of CLR01, there was a marked reduction in the brain load of amyloid plaques and neurofibrillary tangles, the two hallmark pathological lesions in Alzheimer’s disease. The treatment also caused a significant reduction in brain inflammation, which is part of the disease process in Alzheimer’s. There were no side effects associated with the treatment (Attar et al., 2012). A similar reduction of amyloid plaques by CLR01 treatment also was demonstrated in a rat model of Alzheimer’s disease (Malik et al., 2018).
In 2014, we tested the safety and pharmacology of CLR01 and found that the compound was safe in mice at doses 250-times higher than those showing the therapeutic effect in the Alzheimer’s disease mouse model. At the high dose used in the safety study, there were no behavioral or histological findings indicating toxicity. Blood analysis did not find any signs of toxicity. The only significant difference between the mice that received CLR01 and those that received a placebo was approximately 40% reduction in blood cholesterol in the CLR01-treated mice. Additional in-vitro studies demonstrated that CLR01 suppressed the formation of specific Aβ oligomers associated with Alzheimer’s disease (collaboration with the Bowers group, UC Santa Barabra) and protected membranes from disruption by Aβ42 (collaboration with the Jelinek group, Ben Gurion University, Beer Sheva, Israel). More recently, we tested the effect of CLR01 on Aβ42 aggregation and toxicity in the presence of Zn2+, one of several transition-metal ions that had been shown to increase Aβ toxicity. Surprisingly, we found that CLR01 inhibited the toxicity of Aβ42 in the presence of Zn2+ more potently than in the absence of Zn2+, presumably because it can bind both the protein and the metal ion (Mason, Hurst et al., 2020).
The other culprit in Alzheimer’s disease is the protein tau, which forms toxic neurofibrillary tangles in the brain of patients. Tau aggregation also characterizes other diseases, such as frontotemporal dementia, progressive supranuclear palsy, and others (see amyloidoses), together called tauopathies. In collaboration with the Smet-Nocca group at the University of Lille 1, France, we found that CLR01 was an effective inhibitor of tau aggregation and cell-to-cell spreading of aggregated tau (Despres, Di et al., 2019), a mechanism by which Alzheimer’s disease and other tauopathies spread through the brain. Interestingly, NMR experiments by the Smet-Nocca group showed that although there are over 40 potential binding sites for CLR01 in tau, the main binding region was the microtubule-binding domain, which is the most aggregation-prone region in tau. These findings were confirmed separately by the Loo group at UCLA using top-down mass spectrometry (Nshanian et al., 2018). In contrast, CLR01 had little effect on small oligomers formed by recombinant tau (all 6 isoforms) in vitro (Shahpasand-Kroner et al., Prot. Sci. 2022). However, in a study using a mouse model of pure tauopathy, without the involvement of Aβ, we found that CLR01 administered for 35 days protected the mice against deterioration of muscle strength and the development of a disinhibition-like behavior, a typical symptom of frontotemporal dementia. This protection correlated with a significant reduction in hyperphosphorylated, oligomeric, and aggregated forms of tau in the brain of the mice, and a decrease in tau “seeds” that spread the disease throughout the brain. The treatment also significantly reduced neuroinflammation (Di, Siddique et al., Alzheimer’s Res. Ther., 2021). These findings suggested that the tau oligomers formed in the mouse brain were different from those studied in vitro.
Parkinson’s-disease and other synucleinopathies: The first demonstration of the therapeutic effect of CLR01 in a living organism was in a collaborative study with Jeff Bronstein’s group at UCLA using zebrafish embryos genetically engineered to express the human protein α-synuclein, which aggregates and kills dopamine-producing cells in the brain of people who have Parkinson’s disease. Our in vitro characterization of the interaction between CLR01 and α-synuclein showed that CLR01 not only prevented aggregation of α-synuclein but also dissociated pre-formed aggregates (Prabhudesai et al., 2012). The mechanism by which CLR01 inhibited α-synuclein in vitro was explored by the Lapidus group at Michigan State University who found that the molecular tweezer affected the rate of conformational remodeling of α-synuclein and shifted it outside the aggregation range (Acharya et al., 2014). In another zebrafish study by the Bronstein group, CLR01 was found also to protect zebrafish from the neurotoxicity of the pesticide Ziram (Lulla et al. 2016), demonstrating that it could be effective also against environmentally induced Parkinson’s disease. Later, in collaboration with the Chesselet group at UCLA, CLR01 also was shown to ameliorate motor deficits and reduce the accumulation of soluble α-synuclein in the striatum, in a mouse model of Parkinson’s disease expressing human α-synuclein in the brain (Richter et al., 2017).
In a mouse model of the rare synucleinopathy, multiple system atrophy, treatment with 1 mg/Kg CLR01 for 28 days, done in collaboration with Nadia Stefanova‘s group at the Medical University of Innsbruck, led to normalization of an anxiety-like phenotype, in correlation with significant reduction in several pathological forms of α-syn in the brain (Herrera-Vaquero, Bouquio et al., 2019). More recently, in a collaborative study with the Wade-Martins group at Oxford University, CLR01 was found to inhibit intracellular α-synuclein oligomerization with half-maximal concentration (IC50) = 85.4 nM and decreased α-synuclein aggregation in induced pluripotent stem cell-derived dopaminergic neurons treated with Lewy-body extracts from postmortem Parkinson’s-disease brain. CLR01 also improved motor deficits in correlation with reduced α-synuclein pathology in the brain of transgenic mice expressing human, wild-type α-synuclein when administered subcutaneously twice a week at 0.14 mg/Kg . In the same study, CLR01 also was shown to and to protect wild-type mice that received intrastriatal injections of either aggregated recombinant α-syn fibrils or patient-derived Lewy-body extracts (Bengoa-Vergniory et al., 2020).
Interestingly, synuclein accumulation has been found in a lamprey model of spinal-cord injury leading us to ask whether CLR01 could ameliorate neuronal death following spinal-cord injury in these animals. In collaboration with the Morgan group at Marine Biological Laboratories, we found that the compound effectively reduced synuclein aggregation in the lampreys and protected the affected neurons from degeneration (Fogerson et al., 2016).
Other diseases
TTR amyloidosis: The therapeutic effect of CLR01 against TTR amyloidosis was demonstrated in a collaborative study with the Almeida group at the University of Porto, Portugal, who used a mouse model of Familial Amyloidotic Polyneuropathy (FAP), a genetic disease caused by mutations in the gene encoding the carrier protein transthyretin (TTR). Treatment with CLR01 led to a significant reduction in TTR deposition in the affected tissues, with a concomitant decrease in disease markers, including apoptosis (programmed cell death), cellular stress, and protein oxidation.
Desmin-related cardiomyopathy: The group of the late Jeffrey Robbins at Cincinnati Children’s Hospital used CLR01 in cell-culture and mouse models of desmin-related cardiomyopathy, a rare proteinopathy that can be caused by mutations in several proteins. In their model, the disease phenotype was caused by a mutation in the protein αB-crystalline, which causes its aggregation into cytotoxic oligomers and aggregates that kill heart muscle cells. CLR01 treatment led to a significant reduction in the aggregation of the mutant protein and its toxicity (Xu et al., 2017), suggesting that it could be an effective therapeutic agent for this type of heart disease.
Type-2 diabetes: CLR01 prevented aggregation of islet amyloid polypeptide (IAPP), which is associated with degeneration of pancreatic β-cells in type-2 diabetes, at very low concentrations (Lopes et al., 2015).
Cancer: CLR01 prevented the formation of toxic aggregates of the protein p53, which are associated with certain types of cancer (Herzog et al., 2015; collaboration with the Segal group, Tel Aviv University, Israel).
Huntington’s disease: CLR01 inhibited the aggregation of huntingtin exon 1 containing an expanded polyglutamine repeat both in the test tube and in living cells (Vöpel et al., 2017; collaboration with the Ebbinghaus group, then at Ruhr-University, Bochum, and Sanchez-Garcia group, then at Max Planck Institute, Mülheim, Germany).
Amyotrophic lateral sclerosis: CLR01 was found to inhibit the aggregation of several disease-causing variants of superoxide dismutase 1 (SOD1). Daily injection of the molecular tweezer also reduced significantly the accumulation of misfolded SOD1 in the G93A-SOD1 mouse model of ALS (collaboration with Martina Wiedau, UCLA).
Mucopolysaccharidoses: Mucopolysaccharidoses (MPS) are a group of lysosomal-storage diseases, in which mutations in genes encoding lysosomal enzymes lead to formation of defective enzymes, failure of lysosomal degradation and accumulation (storage) of the undigested products. In MPS, the defective enzymes are those responsible for degradation of glycosaminoglycans. Alessandro Fraldi, Federico II University, Naples, Italy, demonstrated in a mouse model of MPS IIIA that in addition to glycosaminoglycans, multiple amyloidogenic proteins, including α-synuclein, tau, Aβ, and prion protein accumulate and aggregate as secondary storage in the lysosomes of neurons in the brain. Following these findings, his group treated the same mice with CLR01, injected daily, and demonstrated that the treatment caused a significant reduction in the accumulation of the aggregated proteins, restored the autophagy-lysosomal pathway function, and improved memory deficits in these mice (Monaco et al., 2020).
Anti-viral activity: In a different direction, CLR01 was found by the Münch group (Ulm University, Germany) and Shorter group (University of Pennsylvania) to disrupt the formation of amyloid fibrils in semen, which are known to enhance HIV infection, thereby reducing infection of target cells by the HIV virus. Surprisingly, CLR01 also attacked the virus itself by destroying its envelope. The study showed that CLR01 was also effective at directly disrupting other enveloped viruses, including hepatitis C virus, human cytomegalovirus, and herpes simplex virus. These results suggested that CLR01 might also be effective against many other enveloped viruses including flu and Ebola. Indeed, follow-up investigations demonstrated the inhibitory activity of CLR01 against Ebola, Zika, SARS-Cov-2, and several other enveloped viruses and deciphered the mechanism by which molecular tweezers disrupt selectively the viral membrane without affecting mammalian cell membranes (Röcker et al., 2018, Weil et al, 2020).
Bacterial biofilm: A study by the Jelinek group at Ben Gurion University of the Negev, Beer Sheva, Israel, and several other groups in Israel, Germany, and the US has shown that the molecular tweezers, CLR01 and CLR05, were effective disruptors of bacterial biofilm, in particular of Staphylococcus Aureus. Biofilms are a “mesh” of proteins, sugar molecules, and nucleic acids bacteria use to protect themselves from the host defense mechanisms and from antibiotics. They often cause persistent bacterial infections and reduce the efficacy of antibiotics. Methicillin-resistant Staphylococcus aureus (MRSA) in particular, is responsible for several difficult-to-treat infections in humans. The new results suggest that molecular tweezers could be developed as effective drugs against bacterial biofilm.